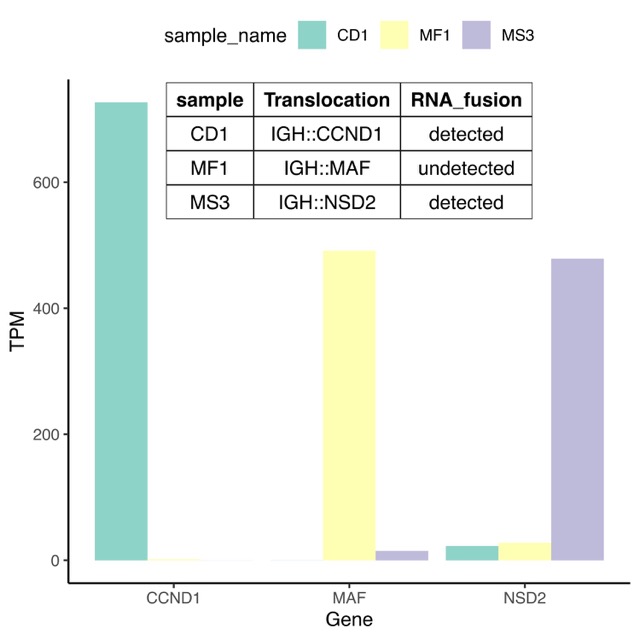
3. Use case II: Fusion genes detection from multiple myeloma patient RNA-seq#
3.1. Background#
Clinical Applications of RNA-Seq in Diagnostic Testing
RNA sequencing (RNA-Seq) is a high-throughput transcriptome profiling technology that enables comprehensive analysis of gene expression, splicing variants, fusion events, and novel transcripts. In clinical diagnostics, it serves as a powerful tool for:
∙ Cancer Subtyping: Identifying tumor-specific gene expression signatures, fusion genes (e.g., BCR-ABL1), and aberrant splicing events to guide targeted therapies.
∙ Rare Disease Diagnosis: Detecting dysregulated pathways and aberrant expression in Mendelian disorders where DNA-based tests are inconclusive.
∙ Infectious Disease Characterization: Profiling host-pathogen interactions and pathogen expression in complex infections.
∙ Biomarker Discovery: Validating expression-based biomarkers for disease monitoring and treatment response.
3.2. Setup a project folder#
Note
Before starting the analysis, please ensure that you have set up the analysis environment using the build_conda_env.sh script.
Create a folder named project/CGGA_WES in your home directory and activate the Clindet conda environment.
mkdir -p ~/projects/MM_RNA
cd ~/projects/MM_RNA
conda activate clindet
3.3. Download data and#
Download Multiple myeloma and COLO829 cellline RNA-seq data from the SRA database using wget and prepare the sample information file, make sure fastq-dump are in in $PATH (if don’t install it first)
cd ~/projects/MM_RNA
mkdir -p data && cd data
## Methods one multiple myeloma RNA-seq data
wget -q -c -O A26.11 https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR12099713/SRR12099713
wget -q -c -O A27.19 https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR12099714/SRR12099714
wget -q -c -O A28.15 https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR12099715/SRR12099715
fastq-dump --gzip -O /public/ClinicalExam/lj_sih/projects/project_clindet/data/GSE153380 --split-3 ./A26.11
fastq-dump --gzip -O /public/ClinicalExam/lj_sih/projects/project_clindet/data/GSE153380 --split-3 ./A27.19
fastq-dump --gzip -O /public/ClinicalExam/lj_sih/projects/project_clindet/data/GSE153380 --split-3 ./A28.15
Next, create a CSV file named pipe_rna.csv in the ~/projects/MM_RNA directory with the following content:
Tumor_R1_file_path,Tumor_R2_file_path,Normal_R1_file_path,Normal_R2_file_path,Sample_name,Target_file_bed,Project
~/projects/MM_RNA/data/A26.11_1.fastq.gz,~/projects/MM_RNA/data/A26.11_2.fastq.gz,MF1
~/projects/MM_RNA/data/A27.19_1.fastq.gz,~/projects/MM_RNA/data/A27.19_2.fastq.gz,MS3
~/projects/MM_RNA/data/A28.15_1.fastq.gz,~/projects/MM_RNA/data/A28.15_2.fastq.gz,CD1
3.4. Write an Snakemake file from template#
For this project, modify the sample sheet and create a new Snakemake file named snake_rna.smk (see below). Set the following parameters in the Snakemake file:
configfile (str): config file for softwares and resource parameters.
stage (list): analysis steps. avaiable options:
['RSEM','arriba','TRUST4','samlom','kallisto']
3.5. write Snakemake file#
For this project, we need change the sample sheet info.
Tip
import pandas as pd
samples_info = pd.read_csv('./pipe_rna.csv',index_col='Sample_name')
unpaired_samples = samples_info.loc[pd.isna(samples_info['R2_file_path'])].index.tolist()
paired_samples = samples_info.loc[~pd.isna(samples_info['R1_file_path'])].index.tolist()
configfile: "/public/ClinicalExam/lj_sih/projects/project_clindet/build_log/config.yaml"
import os
if not os.path.exists("logs/slurm"):
os.makedirs("logs/slurm")
groups = ['NC','T']
stages = ['RSEM','arriba','TRUST4','samlom','kallisto']
caller_list = ['sentieon_anno_rnaedit','Mutect2_filter']
project = 'RNA'
genome_version = 'b37'
rna_res_list = [
##### for isoform expression ######
"{project}/{genome_version}/results/summary/RSEM/{sample}/{sample}.genes.results" if 'RSEM' in stages else None,
##### ka
"{project}/{genome_version}/results/summary/kallisto/{sample}/abundance.tsv" if 'kallisto' in stages else None,
##### for fusion gene detection #####
"{project}/{genome_version}/results/fusion/{sample}_arriba_fusion.tsv" if 'arriba' in stages else None,
##### for TRUST4 immu analysis #####
"{project}/{genome_version}/results/IG/TRUST4/{sample}_report.tsv" if 'TRUST4' in stages else None,
#### Case report #####
]
rna_res_list = list(filter(None, rna_res_list))
rule all:
input:
## paired sample
expand(rna_res_list,
# sample = paired_samples,
sample = ['CD1','COLO829'],
project = project,
genome_version = genome_version
),
##### Modules #####
include: "workflow/RNA/Snakefile"
3.6. Run clindet#
There is two way you can run clindet
run on a local server
submit to HPC through slurm
3.6.1. Run on local node#
nohup snakemake -j 30 --printshellcmds -s snake_rna.smk \
--use-singularity --singularity-args "--bind /your/home/path:/your/home/path" \
--latency-wait 300 --use-conda >> rna.log
3.6.2. Submit to HPC use slurm#
we provide a slurm config.yaml under clindet/workflow/config_slurm folder.
nohup snakemake --profile workflow/config_slurm \
-j 30 --printshellcmds -s snake_rna.smk --use-singularity \
--singularity-args "--bind /your/home/path:/your/home/path" \
--latency-wait 300 --use-conda >> rna.log
3.6.3. Output#
3.7. Results#